


Clinical trials - participation
Compliance rules
0Changes From Previous Versions
No history available so far!
1General information
Clinical trials are conducted for the benefit of patients with the aim of further improving current treatment options for a disease. Participation in such studies gives you the opportunity to be treated with modern and new, perhaps more effective therapies. With your participation, you also make an important contribution to the further optimization of existing therapy concepts. During your participation in the study, you will be intensively monitored and examined. Clinical trials can also be associated with additional burdens, as you may have to attend regular appointments that are necessary to monitor your health status beyond treatment. Depending on the study, patient surveys are also often conducted, so that in addition to the medical aspect (treatment success), the psychological or psycho-oncological aspect is also considered (living conditions / quality of life, etc.). After all the treatment investigated may be superior or inferior to the otherwise usual standard of care and obtaining this information is an important aspect of clinical studies, thus facilitating scientific progress and improving therapies.
1.1Important components of a study
1.1.1The protocol
The planning of a clinical study usually begins with the preparation of a study protocol. In the study protocol, the clinical course of the study is described, the study endpoints ("objectives"), the method for data evaluation, the measured values to be collected (laboratory values, physical examinations, etc.), and the inclusion and exclusion criteria are precisely defined. This guarantees a comparable, safe treatment of all patients participating in the study and enables a reliable statistical evaluation of the treatment results after completion of the study.
1.1.1.1Goals of the study
The objectives of a clinical trial are also referred to as the "endpoints" of the trial. There are primary and secondary endpoints in studies. The primary endpoint is the predetermined, primary goal of a clinical trial. In large trials this is often defined as “improvement in overall survival”. However, response to treatment or reduction in side effects can also be a primary endpoint. The secondary endpoint, describes secondary objectives of a clinical trial. This secondary endpoint can additionally describe a specific therapeutic effect or the safety of the applied therapy but is not in itself proof of the efficacy or superiority of a new therapy.
1.1.1.2Inclusion and exclusion criteria
These criteria usually include conditions that ensure the safe conduct of the study. Only if a patient fulfills all inclusion criteria, no exclusion criteria are present, and his or her written consent to participate in the study has been obtained, the patient can be included in the study.
1.1.2The patient information
Every patient who is treated within a study receives comprehensive information from the treating physicians prior to inclusion in the study. Part of this information is the patient information which is given to you in written form. This document contains all the information that is important for patients to be able to assess the scope of the study and the associated risks. It includes important details such as the treatment plan, its course, the examinations within the study, side effects of the therapy as well as legal information on insurance and data protection. The rights and obligations for patients participating in the study are also described. The content of the patient information may differ from study to study.
TIP: After you have received the patient information leaflet and the explanation from your physician, you do not have to decide immediately for or against participation in the study. It is customary to allow a timeperiod of at least 24 hours to consider a participation.
1.1.3The Ethics Committee
Before the first patients are included in a study, approval must also be obtained from the responsible ethics committee. The ethics committee is an independent and superior control body. In Germany there is at least one ethics committee in each federal state. Its members belong to different professional groups. In most cases, they are physicians, lawyers, natural scientists, theologians or philosophers, and laypersons. The composition is determined by state law and can vary from state to state. The respective ethics committee checks whether all protective regulations for conducting studies are observed and whether, for example, the possible benefits of the study outweigh the possible risks. The content of the patient information is also subject to review.
1.1.4Data evaluation
During therapy, patient safety and treatment efficacy data are regularly monitored by an independent group of experts during an ongoing clinical trial. This group is called the “Data Monitoring Committee” or “Data Safety Monitoring Board”. After completion of therapy, the collected data and measurements are evaluated using statistical analysis methods with respect to the primary and secondary endpoints. These analyses are supervised by experienced biostatisticians. After completion of the data analysis, the study results are usually published in the medical press.
1.2Procedure of a clinical trial
In the following figure, the individual components of the process of participation in a clinical trial are shown again for a better overview.
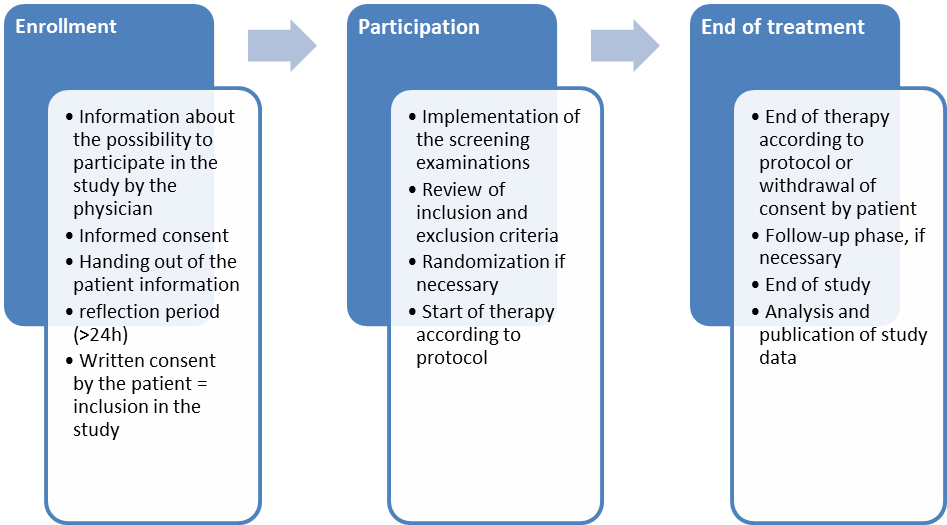
1.3Study types
A distinction is made between interventional (Chapter 1.3.1) and non-interventional (Chapter 1.3.2) studies. While non-interventional studies are often observational in character, intervention studies actively implement a treatment as a planned and targeted measure (intervention).
Below we would like to give you a brief overview of the different types of clinical trials.
1.3.1Interventional studies
There are two forms of intervention studies: the randomized and the non-randomized controlled study. In the randomized form, patients are divided into groups and randomly assigned to the individual therapies to be compared in a predetermined ratio. For example, in a two-arm randomized trial, if randomization or allocation is 1:1, this means that the same number of patients are randomly allocated to both groups (arms). This allocation can`t be influenced by the physician or patient.
The Federal Institute for Drugs and Medical Devices (BfArM) or the Paul Ehrlich Institute (PEI) are responsible for the approval of clinical intervention studies in Germany. A distinction is made between four phases in these studies:
1.3.1.1Phase I study
In Phase I clinical trials, the study is conducted in a very small number of healthy study participants to determine the tolerability and safety of the new drug.
1.3.1.2Phase II study
Based on the results of the Phase I trial, a Phase II trial will then be conducted in a small patient population to determine the optimal dosage of the drug and to collect initial data on the efficacy and tolerability of the drug in patients.
1.3.1.3Phase III study
In the next step, the phase III trial is conducted on a large patient population to demonstrate the efficacy of the new therapeutic procedure. Phase III trials are sometimes also referred to as pivotal trials, as the results of the trial can determine whether a new drug receives approval (from the BfArM) to treat the disease under investigation.
Phase III studies are often conducted as randomized controlled trials or comparative studies. This means that upon trial enrollment, the participants are divided into different groups and the treatment results of the different groups are compared with each other after completion of the study. For example, while one group receives the previous standard therapy and forms the so-called “control group”, the other group is treated with the new therapeutic approach and forms the treatment group. The study participants are randomly assigned to the individual treatment groups. Neither physicians nor patients have any influence on the allocation to the respective treatment group of the study. At the beginning of such trials it is not known which treatment approach is the better one. As soon as a better/worse effect becomes apparent, such studies are stopped and all study patients then receive the better therapy.
1.3.1.4Phase IV study
In phase IV studies, the study participants receive a drug that has already been approved. They are conducted, for example, to specifically investigate the efficacy of the drug again in a larger group of patients under everyday conditions or to identify rare side effects. This is sometimes important; because in the registration studies the administration of the drug is restricted to a certain group of people (e.g. older and sicker patients are often excluded).
1.3.1.5Therapy Optimization Studies
Therapy optimization studies aim to improve a therapy already in use in order to increase the prospects of cure, improve the patient's quality of life, or reduce the long-term consequences of cancer therapy. Therapy optimization studies are often conducted by study groups that belong, e.g. to the competence networks listed in chapter 3.
1.3.2Non-interventional studies
In some situations, controlled and randomized studies are not possible because the requirements for this type of study are not met. The reasons for this could be, for example, that there are too few patients with a certain disease: Thus the group of patients would be too small to be able to make a reliable statement about the benefit of the therapy in a study. Another reason may be of ethical nature: For example, if one group in a randomized trial would have an obvious disadvantage in treatment or would require tissue-injuring (invasive) therapy such as surgery for comparison. Non-interventional studies do not provide an immediate benefit to the study participant itself. However, they can help to further optimize existing therapy concepts.
Non-interventional studies include cross-sectional and cohort studies as well as case-control studies:
1.3.2.1Cross-sectional study
Cross-sectional studies are also called “prevalence studies” and describe the distribution of, for example, a disease in the population at a specific point in time.
1.3.2.2Cohort study
In a cohort study, groups of patients (cohort) who have received a specific therapy (intervention) are followed up over a longer period of time and followed up with regard to the response to therapy. These can be both prospective and retrospective studies.
1.3.2.3Case-control study
If the results of the treatment success are collected and evaluated retrospectively, they are often referred to as case-control studies. They also allow a comparison between previously defined groups, e.g. with regard to the presence of risk factors in a group of patients with a certain disease ("cases") and a group of patients without this disease ("controls").
1.3.2.3.1Good to know
What are my rights and obligations as a participant in a clinical trial?
Patients who are well informed and know their rights can face doctors, health insurers and pharmacists as equal partners.
Before you give your written consent to participate in a clinical trial, you have the right to be fully informed about the content and purpose, the procedure, and the risks and benefits of the clinical trial. For this purpose, you will receive both a patient information form and an informed consent talk with your physician. If you decide to participate in a clinical study, you should comply with the physician's recommendations and the examination times prescribed in the study. Your reliability as a participant is of particular importance for the validity and successful completion of the study. If you have questions about the effects and side effects of the therapy or notice them during the therapy, please always discuss these with the study physicians. You have the right to discontinue participation in a study at any time. If there is any news worth knowing within the study during your participation, you will also be informed about it.
Will I suffer any disadvantages if I withdraw my consent to participate in the study?
In principle, participation in the study is on a voluntary basis. If you withdraw your consent to participate in the study, your doctors will suggest the best available standard therapy. You may then no longer be able to benefit from potential advantages of the medication used within the study (so-called investigational product) or diagnostic methods, as the study medication may only be used with your consent.
What is a double-blind study?
In a double-blind study, different groups (control group, experimental group) are compared with each other. The study participant is randomly assigned to one of the groups. Neither the study participant nor the study physicians can influence the allocation to the respective study group. Neither physicians nor patients have any knowledge of which group the study participant is in.
What happens to my data?
In the context of clinical studies, patients are informed about the use of their data during the informed consent interview. The data collected in the study are used depending on the type of study and the objective of the study. Frequently, these data are "pseudonymized", i.e. encrypted and processed without mentioning your name or other personal data, so that it is not possible to draw any conclusions about your person. Depending on the study, the patient information will contain precise details of the form in which the collected data will be reported to which institution for which period of time. The scope of this data transfer, as well as the period of use of your data, is also stated for the respective study in the patient information. You can request information about your stored data at any time. You also have the right to have incorrect personal data corrected or deleted and to withdraw your consent to the processing of your personal data at any time. In the event of such a withdrawal of your consent, the data stored up to this point may continue to be used insofar as this is required under the German Medicines Act. The German Medicines Act contains more detailed specifications for the required scope of consent to the collection and use of data.
Am I insurance-covered as a participant in a Phase I-IV clinical trial?
Patients in clinical trials are insured for the duration of the treatment. However, the scope and duration of insurance coverage can be individual for each study. Therefore, detailed information on the respective patient insurances is compiled in the patient information. There, the insurance coverage is described in detail.
2Tips and tricks
Be prepared for the educational interview with the study doctor!
Read the patient information carefully, mark the passages about which you have questions and / or write down the questions and ask them to the study doctor during the information session. In addition, it is helpful to bring a relative or friend with you to the patient information session.
Have the individual treatment steps explained to you in detail and ask if there are any other options. If you do not understand something, ask until everything is clear to you. If you have any doubts or are looking for confirmation, get a second opinion from another doctor or at another clinic.
Inquire about fertility preservation options!
Cancer or the therapy of the disease can damage fertility in some cases. Therefore, even in the context of studies, if there is a desire to have children, the options for fertility preservation should be discussed before starting therapy, so that an option for fertility preservation can be taken if necessary. We have compiled further information for you in the AYAPedia guideline "Fertility and Fertility Preservation".
Ask the study physician if an allowance is provided as part of the study.
Travel expenses incurred in connection with participation in a study are sometimes reimbursed. It may therefore be worth asking specifically about this.
During the study you should also talk to your attending physicians if you have any questions or uncertainties!
The daily routine in the clinic often does not allow your attending physicians to have long patient discussions. The appointments are tightly scheduled, the doctors are often brief and/or appear stressed. This can lead to you not having the confidence to ask necessary questions. Remember that you always have the right to ask any questions that are important to you. It helps if you write down the open questions before the appointment and use the notes as a reminder during the interview.
Another possibility to receive answers to pressing questions can be found with the study centers of the research groups that plan and conduct studies. Many of these groups offer the option of contact and support for open questions about the disease and therapy options. Contact addresses can be found in the following chapter 3 (further information).
3Further information
The German Clinical Trials Registry provides information on all clinical trials in Germany: https://www.drks.de/drks_web/
Cancer Support Blue Guide to Clinical Trials: https://www.krebshilfe.de/informieren/ueber-krebs/infothek/infomaterial-kategorie/die-blauen-ratgeber/
Service of the Medical Center for Quality in Medicine (ÄQZ) with information on clinical trials: https://www.patienten-information.de/kurzinformationen/klinische-studien
Patient rights education: https://www.patientenbeauftragter.de/patientenrechte/
Malignant Lymphoma Competence Network: https://lymphome.de/studien/
Competence Network Leukemias: https://www.kompetenznetz-leukaemie.de/content/studien/
European Patient Academy - The ABC of Drug Development: https://www.eupati.eu/de/
5Disclosure of Potential Conflicts of Interest
according to the rules of the supporting professional societies.
Download
Reference:
Quellenangabe:
Onkopedia-Leitlinien werden kontinuierlich an den Stand des Wissens angepasst. Die jeweils gültige Version, AGB und Nutzungsbedingungen finden Sie unter www.onkopedia.com.
Für die kommerzielle Nutzung wenden Sie sich bitte an onkopedia@dgho.de.
Onkopedia guidelines are continuously adapted to the state of knowledge. The currently valid version, terms of use and general terms and conditions can be found at onkopedia-guidelines.info.
For commercial use, please contact onkopedia@dgho.de.
